
Dans la plupart des usines de l’industrie chimique, c’est par distillation que, le plus souvent, on effectue des séparations physiques. C’est un procédé simple qui s’appuie sur la différence de température d’ébullition, c’est-à-dire la différence de volatilité entre produits à séparer. La séparation est réalisée dans des colonnes à plateaux ou à garnissage, où, plateau après plateau, un équilibre est réalisé successivement entre la vapeur et le liquide de chacun d’entre eux.
Toutefois, quand il s’agit de séparer des produits dont les volatilités sont très proches, disons quelques degrés sur la température d’ébullition, la distillation n’est plus adaptée ou n’est pas économique. Il faut alors recourir à d’autre procédés tels que :
- La distillation extractive qui, grâce à l’ajout d’un solvant augmente artificiellement la différence de volatilité mais qui exige une colonne supplémentaire pour distiller ensuite ce solvant.
- L’extraction liquide/liquide qui réalise, en présence d’un solvant bien choisi, des équilibres entre deux phases liquide immiscibles, l’une contenant majoritairement le produit désiré, l’autre le produit non désiré, également sur plusieurs étages successifs.
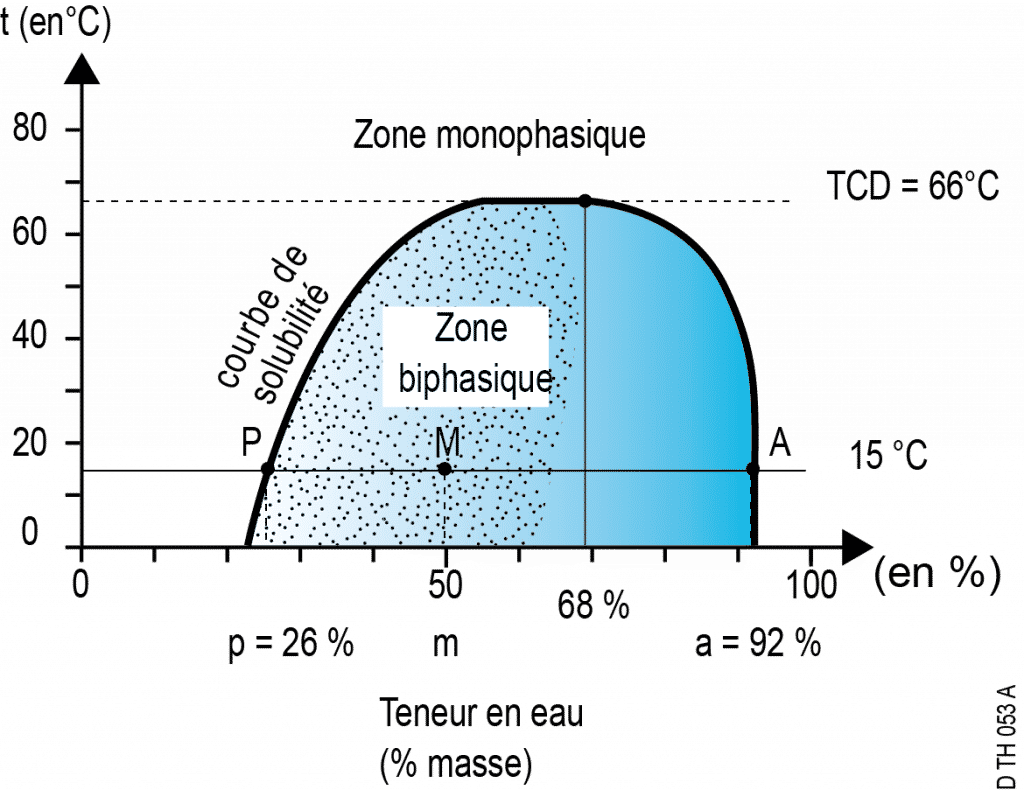
Phénomène d’immiscibilité : exemple du mélange eau/phénol à 15°C

Les segments entre 0 % et P et entre A et 100 % correspondent aux domaines d’existence de mélanges réels, c’est-à-dire de solutions homogènes : on est dans le domaine de miscibilité totale.
Le domaine entre P et A ne correspond pas à des compositions possibles à cette température. En effet, si par exemple on prépare un mélange de 50 % d’eau et 50 % de phénol, le système se sépare spontanément en deux phases dont les compositions en eau sont respectivement égales à 26 % et 92 %. Ce domaine est celui de l’immiscibilité partielle.
- Le point M (situé entre P et A) représente globalement ce mélange ; c’est un point fictif qui symbolise un système constitué de deux phases distinctes.
- Le point P représente la phase riche en phénol contenant un peu d’eau.
- le point A la phase riche en eau contenant un peu de phénol.
À cette température de 15°C, la solubilité maximale de l’eau dans le phénol est de 26 % masse, et celle du phénol dans l’eau est de 8 %.
Influence de la température sur la miscibilité :
- Un accroissement de la température augmente en général les solubilités respectives de deux corps non miscibles.
- Sur un diagramme Température-Composition, les points P et A évoluent sur deux courbes en se rapprochant l’un de l’autre quand la température croît, jusqu’à une limite ou l’immiscibilité disparaît : à cette température appelée « température critique de dissolution » (ou T.C.D.), les courbes de solubilité se rejoignent et les points P et A sont confondus. Dans le cas des mélanges phénol-eau, la T.C.D. est de 66°C et correspond à une composition de 68 % masse en eau.
- Dans la zone biphasique, la répartition entre les deux phase suit la règle des leviers :

PRINCIPE DE L’EXTRACTION LIQUIDE-LIQUIDE À UN ÉTAGE – COEFFICIENT DE PARTAGE
Le principe du procédé d’extraction liquide-liquide est de mettre en contact le mélange liquide de constituants à séparer avec un solvant et de générer ainsi deux phases liquides dans lesquelles les composants de la charge se répartissent. L’une des phases riche en solvant contient des constituants de la charge solubles dans celui-ci. L’autre phase à faible teneur en solvant contient principalement des constituants non solubles dans le solvant d’extraction.
Les différences de comportement vis à vis du solvant permettent de distinguer:
- le soluté, composé qui passe préférentiellement dans le solvant d’extraction
- les autres constituants avec lesquels le soluté était dissous , constituant la charge
C’est la différence de solubilité du soluté vis-à-vis du solvant et de la charge, basée sur les natures chimiques des molécules en présence, qui induit ces différences de comportement.

L’efficacité d’un solvant vis à vis du soluté T recherché est décrite par un coefficient de partage KD .
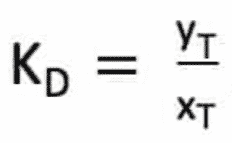
yT est la fraction molaire de T dans la phase lourde, xT la fraction molaire de T dans la phase légère.
On veut, pour un solvant plus lourd que la charge
- Le moins possible de soluté dans la phase légère
- Le moins possible de diluant dans la phase lourde
On regarde donc le pouvoir solvant et la sélectivité :
 Pour faire le design d’une unité d’extraction , il est nécessaire de déterminer :
Pour faire le design d’une unité d’extraction , il est nécessaire de déterminer :
- le nombre d’étages nécessaires aux séparations, pour chaque étape du procédé
- le rapport du volume des phases dans chaque contacteur ;
- les performances des séparations, caractérisées par :
- la teneur en soluté du raffinat ;
- la teneur en soluté de l’extrait avant et après lavage éventuel ;
- les coefficients de séparation des solutés, notamment des impuretés du composé principal à traiter.
A partir des diagrammes d’équilibre, on ne peut accéder par le calcul qu’au nombre d’étages idéaux. Pour pallier cet inconvénient, l’extrapolation des résultats du calcul du nombre d’étages idéaux s’opère de la manière suivante :
- pour les extracteurs à étages individualisés, on définit expérimentalement un rendement pratique de l’étage réel (efficacité de Murphree) que l’on utilise pour obtenir le nombre des étages réels à partir du nombre d’étages idéaux ;
- pour les contacteurs différentiels, on définit, presque toujours expérimentalement, une hauteur équivalente à un plateau théorique ( HEPT), qui, multipliée par le nombre nécessaire d’étages idéaux, donnera la hauteur recherchée de l’équipement.
CRITÈRES DE CHOIX DES SOLVANTS INDUSTRIELS
Le choix judicieux d’un solvant est la clé de la réussite d’un procédé d’extraction. Même si certaines méthodes théoriques peuvent aider à la sélection, il n’existe pas de méthode sûre conduisant au solvant optimal. Le choix d’un solvant ne peut se faire qu’après des expériences de laboratoire mettant en valeur les propriétés qui sont présentées ci-après :
- Coefficient de partage du soluté KD. Il détermine le taux de solvant par rapport à la charge, ce qui impacte la consommation d’énergie (un taux de solvant élevée est souvent la cause d’une consommation d’énergie accrue)
- Sélectivité. Elle traduit la différence de solubilité dans le solvant entre le soluté et le reste de l’alimentation. Plus la sélectivité est grande, plus le nombre d’étages élémentaires successifs est réduit.
- Solubilité du solvant dans l’alimentation. Il est nécessaire que le solvant soit le moins soluble possible dans le diluant, sous peine de nécessiter une opération de séparation supplémentaire de « purification du raffinat ».
- Différence de densité.
Schéma général d’une extraction liquide-liquide
Cette opération industrielle consiste à créer deux phases non miscibles par addition au mélange à séparer d’un solvant sélectif vis à vis des composés de départ. Une fois la séparation de ces 2 phases effectuée, elles doivent être purifiées du solvant qu’elles contiennent. 
-
- La section d’extraction réalise la séparation de la charge et du soluté en présence du solvant : le soluté sort dans l’extrait avec le solvant, le reste de la charge sort dans le raffinat
- La section de purification de l’extrait a pour but de séparer le soluté du solvant. Suivant la nature et les caractéristiques du soluté et du solvant, cette séparation peut faire appel à de très nombreuses techniques : flash successifs, distillation, extraction,. ..
- La section de purification du raffinat est nécessaire chaque fois que le solvant est un peu soluble dans le raffinat. Elle a pour objectif essentiel de récupérer le solvant pour deux raisons principales :
- le solvant dissous dans le raffinat constitue une perte non négligeable, or le solvant est très souvent un produit cher
- le solvant dans le raffinat est souvent une impureté qui gêne les traitements ultérieurs du diluant
- La section de purification du solvant permet d’éliminer de la boucle de solvant les impuretés qui s’y accumulent. Ces impuretés peuvent avoir plusieurs origines :
- Impuretés de la charge très solubles dans le solvant,
- produits de dégradation thermique du solvant qui se forment dans les sections de purification de l’extrait ou du raffinat.
La purification du solvant permet d’éviter une purge de solvant qui serait rapidement onéreuse ou rendue impossible en raison de l’environnement.
MISE EN ŒUVRE DE L’EXTRACTION – extraction continue
Application industrielle :
Il existe différentes manières de réaliser les étages d’extraction nécessaire à l’obtention de la pureté souhaitée.
Dans tous les cas, il est nécessaire d’avoir une succession de zones de mélange et une zone de séparation par décantation
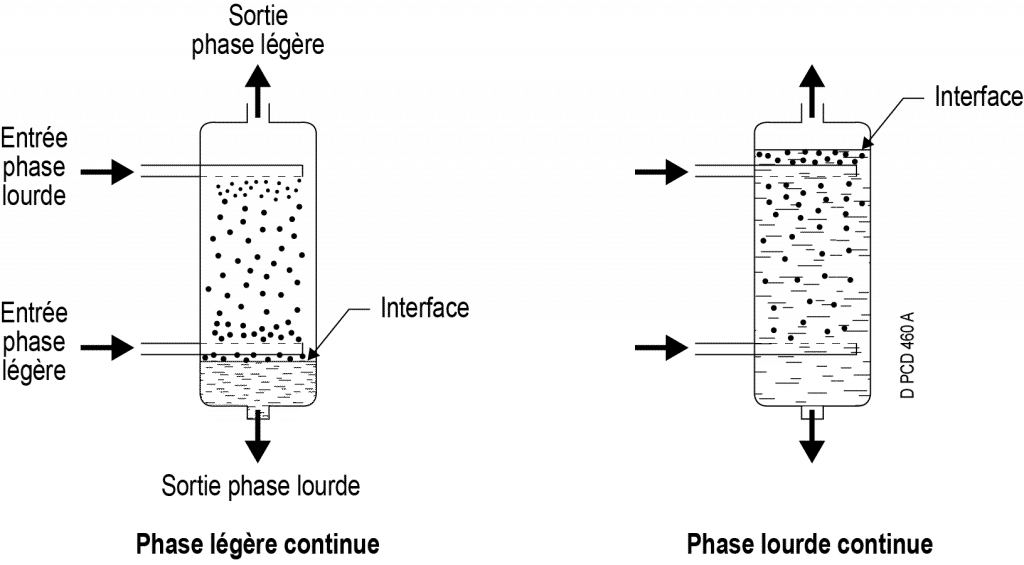
Exemple d’un écoulement en colonne : chacune des phases (lourde et légère) circule par différence de densité.
L’une des deux phases est dispersée, elle s’écoule sous forme de gouttelettes séparées les unes des autres. Selon la position de l’interface, c’est aussi bien la phase lourde qui peut être dispersée (interface en bas) que la phase légère (interface en haut). La phase devra être dispersée au moyen d’un diffuseur, à l’entrée dans l’extracteur ou par tout autre moyen (chicanes, plateaux perforés, agitation, etc.), et cette dispersion doit être entretenue dans l’extracteur (agitations multiples, garnissage, etc.).
L’autre phase, continue, occupe tout le volume non occupé par les gouttes.
Selon la technologie, l’écoulement des 2 phases peut être :
A co-courant
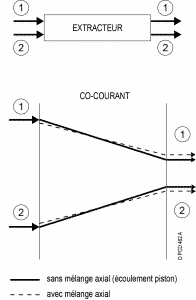
A contre-courant
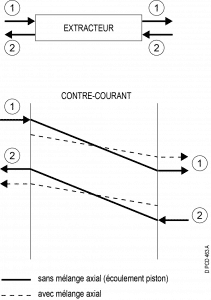
Écoulement globalement à contre-courant, mais les contacts entre phases ont lieu à co-courant.

Courants croisés

Exemples de technologies :
1.Statiques :
 Les plateaux sont de géométrie différente selon que la phase dispersée est la phase lourde ou légère.
Les plateaux sont de géométrie différente selon que la phase dispersée est la phase lourde ou légère.
Les garnissages rencontrés sont du même type que ceux utilisés en distillation.
2.Contacteur à Disques Rotatifs (RDC) :
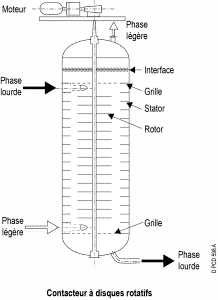
La colonne est séparée en plusieurs tranches par des couronnes horizontales fixes. Dans chaque compartiment, un disque est fixé à un axe central entraîné par un moteur. Cette agitation permet d’améliorer le transfert de matière. La vitesse de rotation variable des disques permet de contrôler la taille des gouttes de la phase dispersée et donc l’efficacité de l’appareil en fonction du débit qu’il doit traiter.
PERFORMANCES DES EXTRACTEURS
- Importance de la taille des gouttes :
- trop grosses : surface de contact entre les deux phases insuffisantes
- trop petites : décantation difficile
- Paramètres de réglage
- la température et donc la viscosité : augmenter la température correspond à diminuer la taille des gouttes
- agitation (par débits élevés ou par agitateur) : favorise le mélange des phases et les petites gouttes
- limite : engorgement. La température, les débits et l’agitation sont limités par le phénomène d’entraînement de phase qui diminue l’efficacité de l’extracteur.