
In the majority of chemicals plants, distillation is the most commonly used technique for physical separation. It is a process that relies on the difference in boiling points, namely the difference in volatility between the products to be separated. Separation takes place in packed or plate columns in which, plate after plate, a balance is reached successively between the steam and liquid in each of them.
However, when separating products with very similar volatility ‒ assuming a few degrees temperature difference in boiling point ‒ distillation is no longer adapted or economical. In such cases, other processes should be considered. These include:
- Extractive distillation which, with the addition of a solvent, artificially increases the difference in volatility but requires an additional column then to distil such solvent.
- Liquid-liquid extraction which, in the presence of a carefully chosen solvent, creates balances between two immiscible liquid phases, one of which contains a majority of the target product and the other of which contains a majority of the non-target product. It also takes place over successive stages.
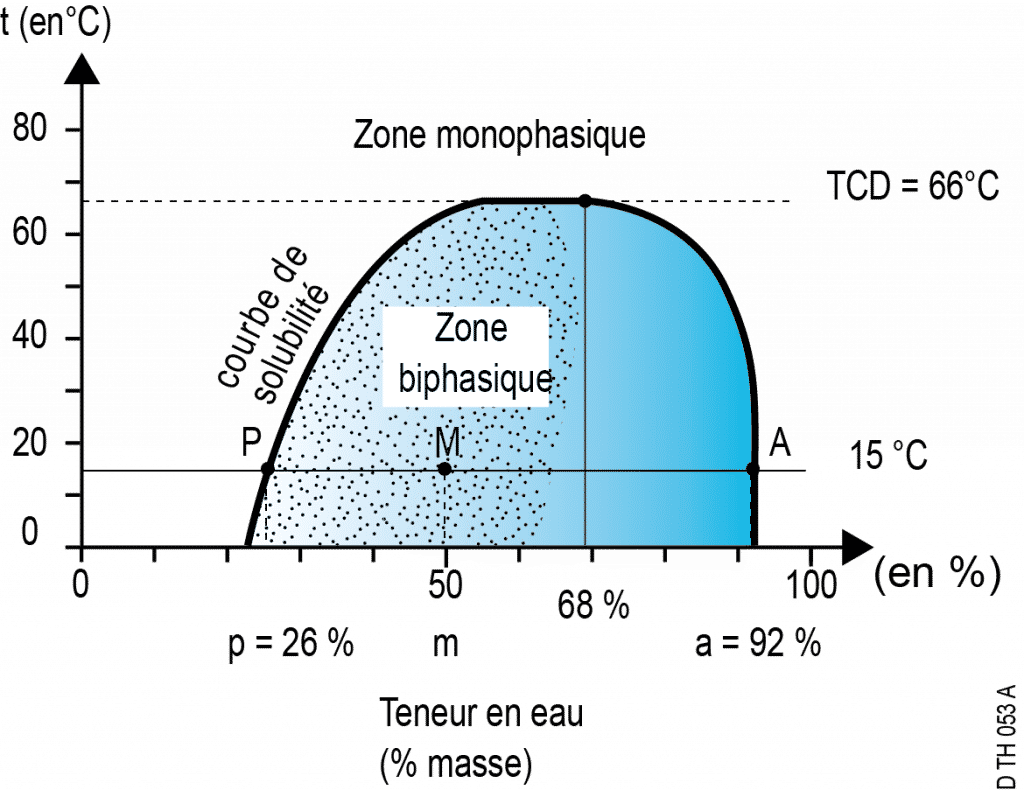
Immiscibility: example of a water/phenol mixture at 15°C

The segments between 0% and P and between A and 100% are the ranges in which real mixtures (i.e. homogeneous solutions) exist: this is the realm of total miscibility.
No compositions are possible at this temperature between P and A. For example, if one prepares a mixture of 50% water and 50% phenol, the system separates spontaneously into two phases whose water compositions are, respectively, 26% and 92%. This is the realm of partial immiscibility.
- Point M (located between P and A) represents this mixture globally; it is a fictitious point which symbolises a system comprised of two distinct phases.
- Point P represents the phenol-rich phase which contains a small amount of water;
- point A represents the water-rich phase which contains a small amount of phenol.
At this temperature of 15°C, the water’s maximum solubility in phenol is 26% mass, while the phenol’s maximum solubility in water is 8%.
Influence of temperature on miscibility:
- An increase in temperature generally raises the respective solubilities of two non-miscible bodies.
- On a temperature composition diagram, points P and A progress along two curves that move closer to one another when the temperature rises ‒ until a limit where immiscibility disappears: at this temperature called “critical solution temperature” (or CST), the solubility curves meet and points P and A are conflated. In the case of water-phenol mixtures, the CST is 66°C and corresponds to a composition with 68% water mass.
- In the binary phase zone, the breakdown between the two phases follows the lever rule:

PRINCIPLE OF SINGLE-STAGE LIQUID-LIQUID EXTRACTION – PARTITION COEFFICIENT
The principle of liquid-liquid extraction is to create contact between the liquid mixture of constituents to separate and a solvent, and thus to generate two liquid phases in which the loaded components are distributed. One of the solvent-rich phases contains the loaded constituents which are soluble in the solvent. The other, low-solvent content phase holds primarily the constituents which are not soluble in the extraction solvent.
The differences in behaviour as regards the solvent make it possible to distinguish:
- the solute, a compound that moves readily into the extraction solvent
- the other constituents with which the solute was dissolved, comprising the load.
These behavioural differences arise from the difference in the solute’s solubility relative to the solvent and the load, based on the chemical natures of the molecules present.

The efficacy of a solvent relative to the solute T sought is described by a partition coefficient KD.
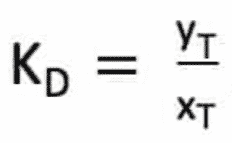
yT is the mole fraction of T in the heavy phase and xT the mole fraction of T in the light phase.
For a solvent heavier than the load, one requires:
- As little solute as possible in the light phase
- As little diluent as possible in the heavy phase
Thus, one considers solvent power and selectivity :

To design an extraction unit, one must determine:
- the number of stages necessary for the separations, for each step of the process;
- the volume ratio of the phases in each contactor;
- the performance of the separations, characterised by:
- the solute content of the raffinate;
- the solute content of the extract before and after any washing;
- the separation factors of the solutes, including the impurities of the main compound to treat.
Calculations based on phase diagrams can define only the ideal number of stages. To compensate for this limitation, the ideal stage number calculations can be extrapolated as follows:
- for stagewise extractors, one experimentally defines a practical yield from the actual stage (Murphree efficiency) which is used to obtain the actual number of stages based on the ideal number of stages;
- for differential contactors, one defines ‒ almost always experimentally ‒ a height equivalent to a theoretical plate (HETP) which, when multiplied by the ideal number of stages, will result in the necessary equipment height.
CRITERIA FOR SELECTING INDUSTRIAL SOLVENTS
Choosing the right solvent is the key to a successful extraction process. Although there are theoretical methods to help with selection, there is no sure method that leads to an optimal solvent. A solvent can only be chosen after laboratory experiments identifying the properties which are described below:
- Partition coefficient for solute KD. This determines the solvent rate relative to the load, which affects energy consumption (a high solvent rate often causes increased energy consumption).
- Selectivity. It translates the difference in solubility in the solvent between the solute and the feed solution. The greater the selectivity, the lower the number of successive individual stages.
- Solubility of the solvent in the feed solution. The solvent must be as insoluble as possible in the diluent, otherwise an additional separation operation will be needed to “purify the raffinate”.
- Difference in density.
Basic flow chart of a liquid-liquid extraction process
This industrial operation consists in creating two non-miscible phases by adding to the mixture to be separated a solvent that is selective relative to the initial compounds. After these two phases have been separated, they must be purified of the solvent they contain.

- The extraction section separates the load and the solute in the presence of the solvent: the solute comes out in the extract with the solvent and the rest of the load comes out in the raffinate.
- The extract purification section is meant to separate the solute from the solvent. Depending on the nature and characteristics of the solute, this separation may involve a wide variety of techniques: successive flashes, distillation, extraction, etc. ..
- The raffinate purification section is needed whenever the solvent is slightly soluble in the raffinate. Its primary purpose is to recover the solvent for two main reasons:
- the amount of solvent dissolved in the raffinate is a significant loss and solvent is often very expensive, and
- the solvent in the raffinate is frequently an impurity that interferes with subsequent diluent treatments.
- The solvent purification section eliminates accumulated impurities from the solvent loop. These impurities can come from several sources:
- impurities in the load which are highly soluble in the solvent, or
- products from the thermal degradation of the solvent which form in the extract or raffinate purification sections.
Solvent purification makes it possible to avoid a solvent purge, which would quickly become expensive or impossible because of the environment.
EXTRACTION IMPLEMENTATION ‒ continuous extraction
Industrial application:
There are different ways to implement the extraction stages necessary to obtain the desired purity.
In all cases, there must be a series of mixing zones and a zone for separating by decantation.
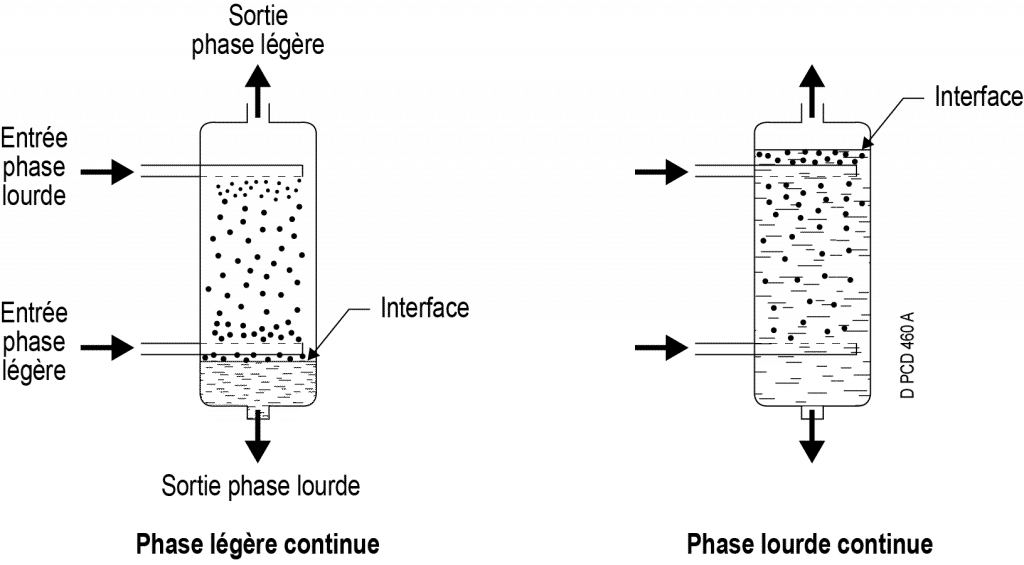
Example of column flow: each of the phases (heavy and light) moves through differences in density.
One of the two phases is dispersed: it flows in the form of droplets which are distinct and separate from one another. Depending on the position of the interface, it is possible for the heavy phase (bottom interface) or the light phase (top interface) to be dispersed. The phase must be dispersed via a diffuser at the entrance to the extractor or by any other means (chicanes, perforated trays, agitation, etc.) and the resulting dispersion must be maintained in the extractor (repeated agitation, packing, etc.).
The other phase ‒ the continuous one ‒ occupies all the remaining volume not occupied by the droplets.
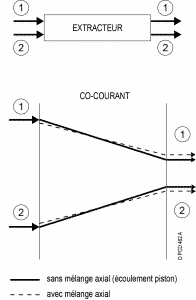
Depending on the technology, there are several types of flow for the two phases :
Concurrent :
Counter-current :
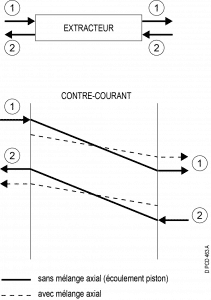
Overall counter-current flow, but concurrent contact between the two phases :

Cross-current :

Sample technologies:
1.Static :

The geometry of the trays varies depending on whether the heavy or light phase is dispersed.
The packing used is the same as what one finds in distillation processes.
2. Rotating disc contactor (RDC) :
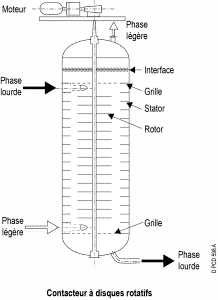
The column is separated into several segments by fixed horizontal rings. In each compartment, a disc is attached to a central motor-driven axle. This agitation improves the transfer of matter. The variable rotation speed of the discs makes it possible to control the size of the droplets in the dispersed phase and, therefore, the efficiency of the device according to the flow it must process.
EXTRACTOR PERFORMANCE
- Importance of droplet size:
- too big: insufficient contact area between the two phases
- too small: decantation is difficult
- Settings
- temperature and thus viscosity: increasing the temperature amounts to decreasing the droplet size
- agitation (by high flow or agitator): promotes the mixing of phases and small droplets
- limitation: clogging. The temperature, flow rates and agitation are limited by the phase entrainment phenomenon which reduces the extractor’s effectiveness.