
La destilación, descubierta probablemente por los persas hace tres mil años, se basa en el hecho de que cuando se calienta un líquido, la fase de vapor que aparece es generalmente más rica en componentes volátiles que el líquido que le dio origen. Durante mucho tiempo, los productores de alcohol eran expertos en destilación.
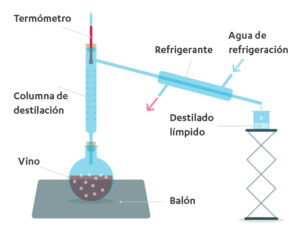
En efecto, al introducir el vino o un puré de frutas fermentado en un matraz, se liberan vapores por calentamiento que, una vez condensados por un enfriador de agua, producen un destilado claro que es el alcohol de 60°.
Gracias al trabajo de los destiladores, complementado principalmente por la investigación de los ingenieros de la industria petrolera, la destilación ocupa un lugar industrial muy importante entre las operaciones de separación.
Profundicemos en la comprensión de esta técnica que se basa, esencialmente, en el conocimiento del equilibrio entre líquido y vapor de la mezcla a separar. Para verlo con claridad, supongamos que deseamos separar por destilación una mezcla binaria de benceno(1) y tolueno(2) disponible a P = 1 atm. Industrialmente, esta separación se realiza en una columna con 19 platos.
La figura siguiente muestra el diagrama dentro de la zona de fases del sistema considerado, calculado bajo presión atmosférica. Este diagrama está formado por la curva de ebullición (límite entre los dominios líquido y bifásico) y la curva de rocío (límite entre las zonas las zonas gaseosa y bifásica). Entre ambas curvas (dentro del husillo), el sistema está en equilibrio líquido-vapor. Para cualquier punto M perteneciente al a la zona bifásica, los puntos L y V, que representan las fases líquida y vapor, se sitúan en las curvas de ebullición y rocío respectivamente. La composición de cada una de las fases puede leerse en el eje de abscisas. A presión atmosférica, la temperatura de ebullición del benceno [Teb(1) = 353 K] es inferior a la del tolueno [Teb(2) = 384 K]. Por tanto, el benceno es más volátil que el tolueno.
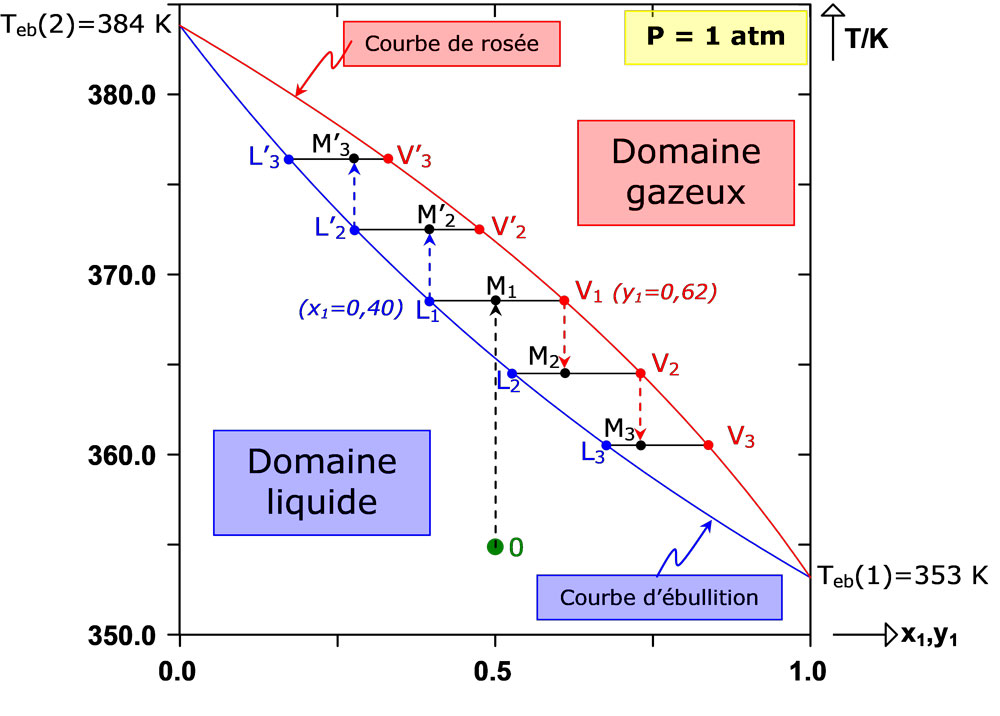
Supongamos que la mezcla a separar, que contiene un 50 % de benceno y un 50 % de tolueno, se almacena en forma líquida en un tanque con una temperatura de T0 = 355 K. El punto que representa el estado del sistema es el punto verde (punto 0) en la figura.
Calentando a T1 = 367 K, se obtiene un sistema bifásico, representado por el punto M1. La fase de vapor (punto V1) contiene entonces un 62 % de benceno. Por tanto, se ha concentrado en el componente más volátil. Asimismo, la fase líquida (punto L1) contiene un 60 % de tolueno, es decir, se ha concentrado en el componente más pesado. Se consiguió una separación parcial de ambos componentes, ya que la mezcla equimolar inicial dio lugar a una fase líquida que contenía un 60 % de tolueno y una fase gaseosa que contenía un 62 % de benceno.
Repitiendo el procedimiento, sería posible obtener ambos componentes con una pureza cercana al 100 %.

Para ello, bastaría con:
1) condensar parcialmente el vapor V1 por enfriamiento para obtener el equilibrio líquido-vapor L2-V2 y luego condensar parcialmente V2 para obtener el equilibrio L3-V3.
Esto generaría fases de vapor (V1, V2, V3, etc.) cada vez más ricas en benceno, es decir, el componente volátil.
Y simultáneamente,
2) vaporizar parcialmente calentando el líquido L1 para obtener el ELV L’2-V’2 y luego vaporizar L’2 para obtener el equilibrio L’3-V’3. Esto generaría fases líquidas (L1, L’2, L’3, etc.) cada vez más ricas en el componente pesado (tolueno).
Desde el punto de vista industrial, una distribución de este tipo sería difícil de concebir, ya que las cantidades de benceno y tolueno producidas serían muy inferiores a las introducidas inicialmente. El diagrama anterior muestra que las corrientes de material L2, L3, V’2 y V’3 se eliminan sistemáticamente del proceso de separación. Para optimizar la operación de separación, basta con reciclar cada corriente líquida L2, L3,…,Li, previamente eliminada, en la etapa de separación de rango (i-1). Del mismo modo, cada corriente de vapor V’2, V’3, … V’i se reciclará a la etapa de rango (i’-1). Esto nos da el diagrama del principio de la destilación industrial continua:

En la práctica, esta sucesión de equilibrios de fase se implementa en una columna de destilación isobárica alimentada continuamente por la mezcla a separar. La columna también tiene dos salidas de material: una en la parte superior de la columna, en el condensador (donde se recupera el destilado, es decir, la mezcla enriquecida en componentes volátiles) y otra en la parte inferior, en la caldera (donde se recupera el residuo, es decir, la mezcla empobrecida en componentes volátiles). El intercambio de material a contracorriente y el intercambio de calor son la consecuencia del retorno a la columna:
1) de una fracción del flujo de líquido del condensador. Este flujo frío y descendente de material es el reflujo de la columna.
2) de una corriente de vapor procedente de la caldera. Es una corriente de material caliente y ascendente.

La parte de la columna situada por encima de la etapa de alimentación se denomina sección de enriquecimiento (o rectificación). La parte de la columna que se encuentra por debajo de esta etapa se denomina sección de agotamiento (o empobrecimiento).
Para garantizar un buen contacto entre el líquido que desciende y el vapor que asciende, la columna está equipada con platos o llena de relleno estructurado.

Sin embargo, al igual que muchas operaciones de transferencia de materiales, el dimensionamiento de una columna de destilación se basa en la noción de bandeja teórica: una bandeja en la que las fases de líquido y vapor estarían en equilibrio termodinámico. Como los tiempos de residencia son limitados, los platos reales de una columna de destilación nunca alcanzan el equilibrio termodinámico. Por lo tanto, en la práctica se necesitan más platos reales que el número teórico de platos estimado a partir del diagrama de fases isobárico. Para tener en cuenta que los fluidos que salen de una bandeja no están en equilibrio, el rendimiento global (EG) de la columna se define como:
EG = número de platos teóricas / número de platos reales
Es una cantidad fácil de medir experimentalmente en una instalación determinada.